
|
第5・6回 「遺伝子の内容を読んだり書き換えることができる」の記録1(遺伝子組み換え実験) |
洗剤などに入っている酵素やチーズの生産に欠かせない「キモシン」と呼ばれる酵素や糖尿病治療薬の「インシュリン」などの医薬品は遺伝子組み換えの技術を使うことで量産が可能になったものです。
遺伝子組み換え大豆やトウモロコシ、トマトなどの遺伝子組み換え作物(GM作物Genetically Modified)は、アメリカで遺伝子組み換えを行っていない作物よりも多く作られるようになっています。
一方、スーパーなどの食品売り場で売っている納豆などには「遺伝子組み換えダイス不使用」と書かれています。最近では「遺伝子組み換え作物を飼料にも使っていません」と表示している牛肉なども目にします。
このように遺伝子組み換え食品の安全性に対する疑問の声も聞かれます
最近では遺伝子組み換えペット(GMペット)も話題になりました。(光るメダカや光るゼブラフィッシュ) 今回の講座ではオワンクラゲの発光タンパク質GFP(Green Fluorescent Protein)の遺伝子をプラスミドを使って大腸菌K12株に導入し、大腸菌が光る様子を観察します。
その中で、 遺伝子が働く仕組み。 遺伝子組み換えの原理。 理解することが目的です。
また、大腸菌などの細菌を扱う方法についても学びます。 その上で遺伝子組み換え技術のプラスの面とマイナスの面について考えてもらいたいと思います。
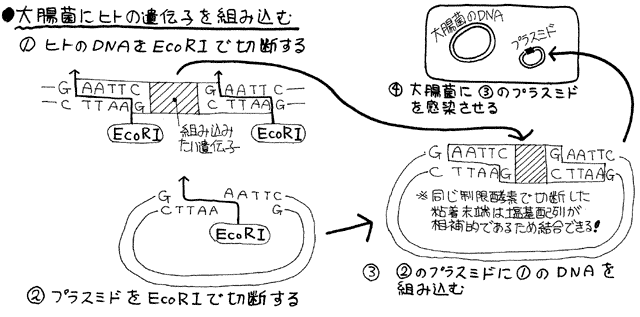
遺伝子組み換えとは、何らかの方法で手に入れた遺伝子(実際にはDNAの断片)を細胞に組み込み、働くようにすることです。
この技術は、動植物の品種改良、医薬品の開発、遺伝病の診断や治療などに利用することが期待されています。
また、生物学や医学、薬学などの研究に欠かせない手段にもなっています。
組み込みたい遺伝子を組み込まれる細胞に運ぶ役割をさせるDNAの断片。大腸菌に組み込む場合はプラスミドがそれ以外ではウイルスがよく使われる
大腸菌などの細菌で染色体とは別に細胞質中にある環状のDNA。染色体の遺伝子とは独立して増殖し、他の細胞へ移動する能力(感染性)を持つ。
この性質を利用し、プラスミドに組み込みたい遺伝子を組み込みプラスミドを細菌に感染させることで遺伝子を細菌に組み込むことができる(プラスミドをベクターとして利用する)。
抗生物質に対する耐性遺伝子を持つものがよく知られている。これらのプラスミドは菌から菌へ次々と感染するので抗生物質に対する耐性は急速に広がる。
遺伝子がタンパク質の殻に入っているだけで細胞を持たない。
単独では自分の複製を作る(自分の子孫を作る)事ができない。生きている細胞に感染し自分の遺伝子を宿主の細胞内で働かせ、自分の複製を作らせる事で増殖する。
一部のウイルスは自分の遺伝子を宿主のDNAの中に組み込んでしまう性質がある。
この性質を利用し、プラスミドを持たない細菌以外の生物に遺伝子を導入するベクターとして利用される。
大腸菌などの細菌に感染する「ウイルス」。細菌がかかる病気の病原体のようなものである。
細菌は自分自身をファージから守るためにファージのDNAを切断する酵素を持っている。これが「制限酵素」である。
制限酵素で切断したDNAの切断箇所は「粘着末端」と呼ばれ図のように段違いに切れている。
制限酵素は特定の塩基配列を認識しその部分を段違いに切断する。
したがって、同じ制限酵素で切断したDNAの断片どうしはリガーゼと呼ばれる酵素によって結合出来るが、異なる種類の制限酵素で切断した断片は結合出来ない。
同じ制限酵素で切断したDNAの粘着末端同士を結合させる酵素。
RNAの遺伝情報をDNAに逆転写する酵素。RNAポリメラーゼ(DNAの遺伝情報をRNAに転写する)の逆の働きをする。
導入したい遺伝子を含むDNAを手に入れます。
直接手に入れにくい場合、逆転写酵素を使ってm-RNAから作ることもできる。塩基配列が明らかであれば人工的に作ることも出来る。
導入したい遺伝子を含むDNAを制限酵素で切断します。
ベクター(運び屋)として使うプラスミドやウイルスのDNAを1と同じ制限酵素で切断します。
2の制限酵素で切断したベクターと1のDNAを混合し、リガーゼと呼ばれる酵素を加え両者を結合させます。
同じ制限酵素で切断したDNAは切断面が相補的になるので、リガーゼで接着することが出来る。 制限酵素がハサミ、リガーゼがのりだと思えばよい。
導入したい遺伝子を結合させたベクターを細胞に感染させます。ベクターが細胞のDNAに組み込まれます。 プラスミドは細胞にはいるだけで働く。ウイルスは細胞のDNAに自分のDNAを組み込んでしまう性質がある。
今回の実験では、感染性のないプラスミド(そのままでは取り込まれない)を使い、ヒートショックと呼ばれる方法で大腸菌に取り込ませます。
遺伝子の導入が成功する確率は低いので、成功した細胞だけを選び出す方法が大切です。
マーカーとしてよく使われるのは特定の抗生物質に耐性を持つ遺伝子です。 GFPの遺伝子もマーカーとして使われます。今回の実験ではアンピシリンと呼ばれる抗生物質の耐性遺伝子をマーカーとして使います。
たとえばペニシリン耐性遺伝子をマーカーとして組み込んだ場合。遺伝子を組み込んだ後、細胞をペニシリンを含む培地で培養すれば、ペニシリンに耐性を持つ遺伝子を組み込まれた細胞だけ生き残ります。
生き残った細胞には目的の遺伝子も組み込まれているはずです。
今回の実験は大腸菌に導入するプラスミドにはGFPと呼ばれる紫外線を当てると緑色の蛍光を発するタンパク質と、アンピシリンと呼ばれる抗生物質に耐性を持たせる遺伝子があらかじめ組み込まれています。
そのために、アンシピリンを含む培地では遺伝子組み換えに成功した大腸菌だけが生き残り、紫外線を当てると緑色の蛍光を発します。
遺伝子が働くためにはDNAの遺伝情報をm-RNAに転写する必要があります。 これは「RNAポリメラーゼ」と呼ばれる酵素の働きによるものです。
RNAポリメラーゼはDNAの上を決まった方向に滑りながらDNAの遺伝情報をm-RNAに転写していきます(RNAポリメラーゼが移動する方向を川の水にたとえDNAの上流・下流と呼びます)。 また、RNAポリメラーゼはDNAのどの部分からも転写を始められるのではなく、DNAの「プロモーター」と呼ばれる塩基配列の部分にしか結合できません。
つまりRNAポリメラーゼはプロモーターから下流に向かって移動しながらDNAの遺伝情報をm-RNAに転写するのです。 したがって、遺伝子の上流部分にプロモーターがなければその遺伝子は働くことができません。またプロモーターにm-RNAが結合できなければその遺伝子は働くことができません。
実際にプロモーターを失ってしまい働くことがない遺伝子が多数存在します。このような機能を持たないDNAの部分をジャンクと呼ぶこともあります。
m-RNAはそのままタンパク質に「翻訳」(m-RNAの遺伝情報にしたがってタンパク質を合成すること)されるわけではありません。
m-RNAにはタンパク質に翻訳される「エキソン」と呼ばれる部分と、タンパク質に翻訳されない「イントロン」と呼ばれる部分があります。
m-RNAのイントロンは切り取られエキソンの部分だけがタンパク質に翻訳されます。この現象を「スプライシング」と呼びます。
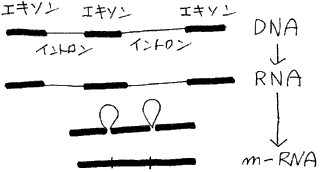
ほとんどの遺伝子はいつでも働いているわけではなく、必要なときにだけ働き、必要がないときには遺伝子は働いていないはずです。
遺伝子には必要なときにだけオンになるスイッチがついています。また、そのスイッチを操作する働きを持つ遺伝子も存在することが明らかになってきました。これを調節遺伝子あるいはDNAの調節領域と呼びます。
遺伝子スイッチのもっとも単純なものはオペロンと呼ばれ、細菌がもっています。
次の図は大腸菌のアラビノースオペロンを示したものです。
大腸菌はグルコース(ブドウ糖)を栄養分として利用していますが、アラビノースが存在するとアラビノースも分解して利用することができます。
大腸菌はアラビノースを分解するアラビノース分解酵素をいつでも作っているのでしょうか。詳しく調べてみると、大腸菌はアラビノースがあるときだけアラビノース分解酵素を作ることがわかります。 つまり、アラビノースがあるときだけアラビノース分解酵素の遺伝子のスイッチがオンになるわけです。
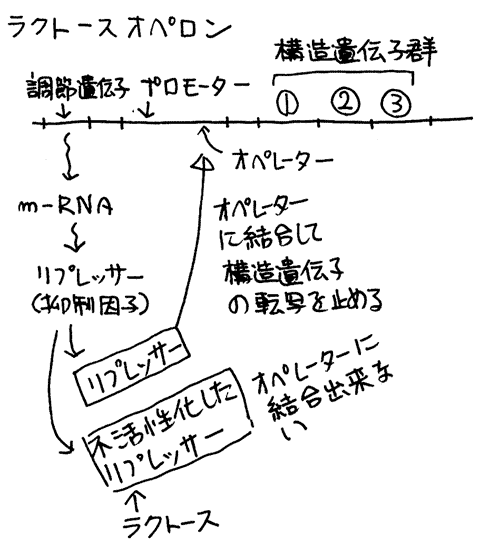
アラビノースを分解するために必要な遺伝子は全部で三種類あります。これらの遺伝子はDNA上に並んで存在し、「構造遺伝子群」と呼ばれます。
そのすぐ上流部分にRNAポリメラーゼが結合するプロモーターがあります。
アラビノースがあるときはこのプロモーターにRNAポリメラーゼが結合しm-RNAが作られます(プロモーターがオン)。
アラビノースがないときはプロモーターの一部にリプレッサー(抑制因子)と呼ばれるタンパク質が結合し、プロモーターにRNAポリメラーゼが結合できなくなります(プロモーターがオフ)。このためにm-RNAが作られなくなります。
リプレッサーが結合するプロモーターの一部分をオペレーターと呼びます。
リプレッサーの遺伝子は調節遺伝子と呼ばれ、プロモーターのさらに上流にあります。
では、アラビノースがあるときはなぜリプレッサーがオペレーターに結合しないのでしょうか。
じつは、アラビノースはリプレッサーに結合しリプレッサーがオペレーターに結合できなくしてしまうのです。
オペレーターに結合しているリプレッサーにもアラビノースは作用しリプレッサーを不活性化します。
図のラクトースオペロンはアラビノースオペロンと同じ仕組みでラクトースがあるときにだけラクトース分解酵素の遺伝子が働きます。
アラビノースなし→リプレッサーがオペレーターに結合→RNAポリメラーゼがプロモーターに結合できない(プロモーターがオフ)→m-RNAが作られない→アラビノース分解に必要な酵素が作られない。
アラビノースあり→リプレッサーが働かない→リプレッサーがオペレーターに結合しない→プロモーターにRNAポリメラーゼが結合する(プロモーターがオン)→m-RNAが作られる→アラビノース分解に必要な酵素が作られる。
アラビノースがないときにはオフになっているアラビノース分解に必要な酵素群の遺伝子(構造遺伝子群)のスイッチ(プロモーター)がアラビノースによってオンになるわけです。
大腸菌はグルコースが豊富にあるときにはアラビノースがあってもほとんどアラビノースを利用しません。 大腸菌はグルコースがあるなしによってもアラビノース分解酵素をコントロールをする仕組みもあわせ持っていることがわかります。
トリプトファンと呼ばれるアミノ酸は大腸菌にとってなくてはならない物質です(ヒトにとってもなくてはならない物質ですが)。 大腸菌はトリプトファンが培地にないときには自分で作ることができます。
しかし、トリプトファンが培地になるときにはわざわざ自分で作らず培地にあるトリプトファンを使います。
大腸菌はトリプトファンがないときはトリプトファンの合成に必要な酵素群が作られ、トリプトファンを合成します。
トリプトファンがあるときにはトリプトファンの合成に必要な酵素群は作られません。
このことは、トリプトファンがトリプトファン合成に必要な酵素群の遺伝子のスイッチをオフにする働きをしていると考えられます(ラクトースと逆です)。
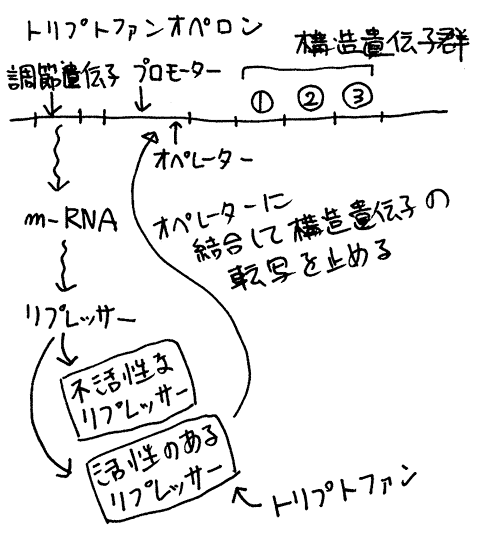
トリプトファンオペロンではラクトースオペロンと同じようにトリプトファンの合成に必要な酵素群の構造遺伝子群の上流にプロモーターがあり、プロモーターの一部分にオペレーターがあります。
異なるのは、調節遺伝子の作るリプレッサーはそのままではオペレーターに結合できないということです。このリプレッサーはトリプトファンと結合してオペレーターに結合できるようになります。
そのため、トリプトファンがないときはリプレッサーはオペレーターに結合せず、プロモーターにRNAポリメラーゼが結合し(プロモーターがオン)、トリプトファンの合成に必要な遺伝子群のm-RNAが作られ、トリプトファンが合成されます。
トリプトファンがあるときにはリプレッサーがオペレーターに結合するため、RNAポリメラーゼがプロモーターに結合できず(プロモーターがオフ)m-RNAが作られず、トリプトファンは合成させません。
つまり、アラビノースオペロンの調節領域をGFPの遺伝子の上流に組み込んだものです。そのため、アラビノースはGFPの遺伝子のプロモーターにあるオペレーターに結合するリプレッサーを不活性化します。
アンピシリン耐性遺伝子は常にオンになっています。
アンピシリン耐性遺伝子はβ−ラクタマーゼという酵素の遺伝子です。この酵素を持つ細菌はアンピシリンを分解できるためアンピシリンを含んだ培地でも生育が可能となります。
オペロンのように遺伝子の発現をコントロールする仕組みはほかにもたくさん見つかっています。 ヒトのような多細胞生物では、大変複雑な仕組みを持つことがわかっています。
すべての細胞は同じ遺伝子を持つにもかかわらず、様々な種類の細胞に分化しているということは、それぞれの細胞で働いている遺伝子が異なることを意味します。
したがって、遺伝子の実際の働きを理解するためには生物が持つ遺伝子の働きを知るだけでは不十分です。それぞれの遺伝子がいつどのようにして働くのかを知る必要があります。 そのためには遺伝子スイッチなどの遺伝子の働きをコントロールするしくみについて理解する必要があります。
細菌やカビなどの微生物は目に見えません。そこで実験中はどのチューブに何が入っているのか常にイメージを持つように心がけてください。
微生物が生育するのに適した養分などを溶かした液を「培地」と呼びます。微生物の種類によってさまざまな培地が考えられています。
大量の微生物が必要なときは、液体の培地に微生物を混ぜて培養します。容器を静置すると微生物が底に沈殿し、酸素や養分が不十分になります。通常は容器を揺らし微生物が沈殿しないようにします。この方法が最も効率よく微生物を培養することが出来ます。
加熱した培地に寒天(アガロース)を溶かし、シャーレの中で冷やして固めると、板状の培地を作ることが出来ます。これを「寒天培地」または単に「プレート」と呼びます。
液体培地の微生物をうんと薄め、全体で微生物が100個でいどになるぐらいの量をプレートにまきます。この液をプレートに薄くのばすと100個の微生物が1個ずつバラバラになってプレートに付着します。もちろん全く目に見えません。
このプレートを保温すると微生物が増殖し、プレートの上に「コロニー」と呼ばれる白っぽい小さな固まりを作ります。コロニーは1個の微生物が分裂して出来たものです。したがって、コロニーの数を数えれば最初にプレートにまいた微生物の数がわかります。この場合は100個のコロニーが出来ているはずです。
1個のコロニーは数十億個の微生物から出来ています。一つのコロニーを作っている微生物は同じ遺伝子を持つクローンです。
最初にまいた微生物の数が多すぎたり、培養時間が長すぎると、いくつかのコロニーがくっついてしまうことがあります。こうなると、異なるクローンが同じコロニーにまざってしまいます。このようなコロニーを実験に使うべきではありません。そうでない1個の微生物から始まっているコロニーを「シングルコロニー」と呼びます。
専門家はコロニーの色や形から微生物の種類を判断します。
基本操作
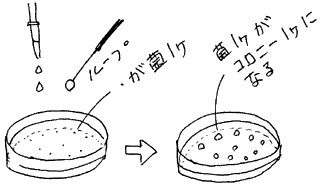
このように微生物実験の基本操作はプレートに微生物をまき、培養し、コロニーの数を数えることです。
今回の実験はBIORAD社のBiotechnology Explorer Kit1を使います。
このキットは10年ほど前からアメリカ販売され、最も広く使われている中学高校向けの遺伝子組み換え実験キットです。 日本でも2002年末から販売が始まりました。
文部科学省でも「教育目的組換えDNA実験指針」が決められ、小中高校の理科室で遺伝子組み換え実験が可能になりました。
「教育目的組換えDNA実験指針」とは、指針で定める宿主とベクターを使い、定められた遺伝子を組み込む場合、指針に定める注意を守ることで小中高校の理科実験室での実験を認めるものです。
アメリカではこのキットに関しては何の規制もなく、デパートで一般向けに販売されており、家庭のキッチンで実験しても大丈夫とされているそうです。
宿主である大腸菌K12株は菌自身が無害なもので、1922年にアメリカのスタンフォード大学で単離され、さまざまな実験に使われその性質がよく分かっています。
この実験に使うK12株は自然のものではなく、DNA修復酵素の遺伝子を取りのぞき、λファージ、F因子(感染性のプラスミド)を取り除く改良を加えてあります。
このため、この菌は遺伝子をほかの菌に渡すことが無く、LB培地の中でないと生育が困難で自然界に放出されても生きることが難しい菌です。またベクターとして使うプラスミドp-GLOには感染性がなく、ほかの大腸菌に移動することがないものです。 生物学的封じ込めB1レベルに相当します。
組み込む遺伝子はGFP(クラゲの発光タンパク質)の遺伝子GFP、およびGFPの調節遺伝子ara-C(アラビノースオペロン)、アンピシリン(スクリーニングに使う抗生物質)耐性遺伝子blaでいずれも害の無いものでいずれも教育目的DNA組換え実験で認められているものです。、
GPLOの構造
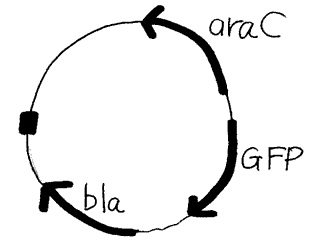
実験指針では上記の条件を満たしている場合、小中高校の理科実験室で実験を行うことが認められています。
遺伝子の名称を示す記号は斜体で書くか下線をつけるます。そうでない場合はその遺伝子がコードしているタンパク質を示すのが習慣です。
以上「教育目的組換えDNA実験指針」より一部改変
微生物を取り扱う場合、二つのことの注意する必要があります。
培地には多くの養分が含まれ微生物が繁殖しやすい環境になっています。特に野生の微生物が混入すると目的の微生物を駆逐してどんどん増えます。
別の微生物が混入すると実験の結果が正しく出ないことになります。 目的以外の微生物が混入することを「コンタミネーション」と呼びます。 専門家は略して「コンタミ」と呼びます。
そのためには「無菌操作」と呼ばれる方法が必要です。無菌操作は野口英世の時代から、微生物を安全に扱い、コンタミを防ぐために工夫されてきました。
今回は特別の設備がない実験室で可能な無菌操作を行います。クリーンベンチやクリーンボックスなどが無くても無菌操作は可能です。
手を石けんでよく洗い、エタノールで消毒します。
実験台の上にエタノールを噴霧し乾く前にペーパータオルでふき取ります。ラボマットの上も同様に消毒します。
マイクロピペット(ピペットマン)の使い方の練習をします。
緑のマイクロチューブに + 青のマイクロチューブに − とマーカーで書き班の番号も書き厚い方のマイクロチューブラックにさしておきます。
BUマイクロチューブ(赤)から形質転換溶液を250μlずつ+−とマークした緑と青のマイクロチューブに分注します。 マイクロピペットP1000を使い、ピペットチップは青を使います。 チューブのふたを閉め薄い方のマイクロチューブラックに深くさし、氷にさしておきます。
p-GLOと呼ばれるこの実験で使うプラスミドには感染性がないために大腸菌に加えても自分で大腸菌に入ることができません。 そのため、ヒートショックと呼ばれる方法で無理やり入れてやる必要があります。
形質転換溶液は0.1mol/lの塩化カルシウムの溶液です。細胞膜を不安定にさせ、プラスミドを取り込ませやすくします。 そのため大腸菌は弱ってしまいます。できるだけ冷却して大腸菌のダメージを少なくします。
植付け用ループを使って、スタータープレートからシングルコロニーを1 つすくいとります。「+チューブ(緑)」の底までループを入れて、人差し指と親指をこすり合わせるようにしてループをまわし、先についている大腸菌を溶液に溶かし入れ、ダマがないようにします。同様に、「−チューブ(青)」にもコロニーを取ります。
正しくはシングルコロニーを一つ取るべきです。しかし、菌の量が少ないとうまくいかないので、コロニーが小さい時は数個のコロニーをすくい取ります。できるだけ2本のチューブに入れる大腸菌の量を同じにします。
大腸菌をチューブに溶かし入れた後は、チューブの先端(溶液の入っている部分)を持たないようにし、すぐにアイスボックスにさします。 暖めないように注意します。
プラスミドの溶液をマイクロピペットP20で10μl「+チューブ(緑)」に加えます(ピペットチップは黄を使います)。
腸菌とよく混ざるようにチップの先端を大腸菌の溶液に入れ何度かゆっくりとピストンを押し、溶液をチップに出し入れします。
チューブは素早く氷に戻します。「−チューブ(青)」には何も入れません。
この操作をピペッティングと呼びます。このときチップの先端を溶液に入れたまま2ndStopまでピペットのピストンを押さないようにします。
+チューブ(緑)には大腸菌とプラスミドが入っています。
−チューブ(青)には大腸菌が入っていますがプラスミドは入っていません。
チューブは10分間氷冷します。その間に4枚のプレートの準備をします。
LB/ は何も含まれていないLB培地です。
LB/amp にはアンピシリンが加えてあります。
LB/amp/ara にはアンピシリンとアラビノースが加えてあります。
LBとLB/ampのうち1枚に /− と書き加えます。
もう一枚のLB/ampとLB/amp/araには /+ と書き加えます。さらに自分の班の番号も書いてください。
+チューブと−チューブをマイクロチューブラックにさしたまま42℃のウォーターバスに正確に50秒浮かべます。このときチューブがマイクロチューブラックに深くさしてあることが重要です。
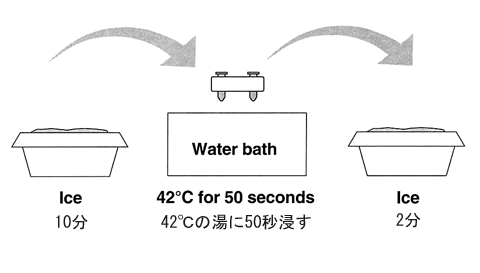
ウォーターバスに入れる時間が90秒になるとプラスミドを取り込む効率は1/10以下になります。正確に素早く行ってください。
急激に温度を上げると細胞膜が柔らかくなりプラスミドが取り込まれます。時間が長いと大腸菌が弱ってしまうのですぐに冷やす必要があります。
プラスミドがヒートショックで取り込まれる仕組みはよく分かっていません。薬品の種類や濃度、温度、時間は経験的に決められたものです。
50秒後 +チューブと−チューブをマイクロチューブラックごと氷に戻します。素早く移動させ確実に冷やすことが大切です。
この操作で不安定になっていた大腸菌が落ち着きます。
2 分間氷冷した後、マイクロチューブラックごとチューブを実験台の上に移します。
+チューブと−チューブに、LB 培地(LBと書いてある透明のチューブ)をマイクロピペットP1000で250 μl 加え、ゆっくりとピペッティングして溶液を混ぜます。次のチューブに移るときにピペットチップは必ず交換します。10 分間室温で放置します。
LB培地はこの大腸菌にとって最も生育しやすい環境です。LB培地を加えることで大腸菌は活動を始め組み込まれた遺伝が働き始めます。
アンピシリン耐性遺伝子が働き始めアンピシリンを分解するβラクタマーゼが作られ始めます。 アンピシリン耐性遺伝子が十分働かないうちにアンピシリンを含む培地にまくと大腸菌は死んでしまいます。
LB/− とLB/amp/− には−チューブの溶液を100μlまきます。

まく前にチューブを軽くたたくが軽く机に打ち付け(タッピング)内容物を十分かき混ぜます。 P200を使ってチューブの溶液を100μl取ります。そのとき十分ピペッティングして内容物を十分かき混ぜてから取るようにします。
大腸菌は放置するとチューブのに沈殿します。かき混ぜないで上澄みだけ取るとほとんど大腸菌が入っていないことになります。
プレートの中央に溶液を滴下し、ループを使って広げます。そのときループを培地と平行になる向きに持ち、培地を傷つけない程度にしっかり素早くこすります。片手でプレートを回しながらループを往復させ(1秒で2往復程度)、プレートを3回転程度させるぐらいがよいでしょう。
プレートを替えるときには必ず新しいループを使います。まきおえたらすぐにフタをします。
同様にして+チューブの溶液を100μlずつ、 LB/amp/+ とLB/amp/ara/+ にまきます。
4枚のプレートに大腸菌をまきおえたら4枚のプレートを逆さまにして重ね、テープで止めます。テープに班名を書いておくとわかりやすいです。
プレートは37℃のインキュベーター(恒温機)に入れ次の日まで培養します。
机の上を片づけエタノールで消毒し手を洗います。
大腸菌をまいた4枚のプレートで大腸菌はどのようなコロニーを作るでしょうか。結果を予想します。
| プレートの表示 | LB培地に加えた成分 | 大腸菌とプラスミド | 結果の予想 |
| LB/− | なし | プラスミドを 組み込んでいない |
生育 する・しない GFPを 作る・作らない |
| LB/amp/− | アンピシリン | プラスミドを 組み込んでいない |
生育 する・しない GFPを 作る・作らない |
| LB/amp/+ | アンピシリン | プラスミドを 組み込んでいない |
生育 する・しない GFPを 作る・作らない |
| LB/amp/ara/+ | アンピシリン アラビノース |
プラスミドを 組み込んでいない |
生育 する・しない GFPを 作る・作らない |
手を石けんでよく洗い、消毒します。
実験台の上も消毒します。
どのような結果が出るか予想してみます。
インキュベーターからプレートを出し、黒のラシャ紙の上に4枚のプレートをLB/− LB/amp/− LB/amp/+ LB/amp/ara/+ の順に並べ、ふたを開けプレートのコロニーを自然光で観察し記録します。
次に暗幕を引き部屋を暗くしてからUVランプで紫外線を照射し、コロニーの様子を観察し記録します。観察時には紫外線カットのゴーグルをつけ、紫外線ランプの光を直接見ないように注意します。
使用するのはブラックランプと呼ばれる市販の長波長紫外線ランプです。有害な短波長の紫外線が出ないため短時間の使用であればゴーグルをつけなくても良いことになっています。
観察と記録が終わったら、LB/amp/+プレートのコロニーの一部にアラビノースの溶液を1滴滴下します。アラビノース溶液が流れないように水平にしたままフタをし、アラビノース溶液を滴下した場所を覚えておくか、フタにマーカーで目印を付けます。
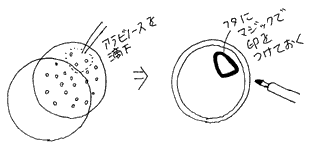
4枚のプレートをひっくり返さずに重ね、テープで止め、インキュベーターに入れ数時間培養をします。 LB/amp/+プレート がどうなるか結果を予想します。
LB/amp/+プレートの状態 |
|
培養している間に、DNAフィンガープリントの実験を行います。この実験を始める前に実験台と手を消毒し、手を洗います。
数時間(1時間以上)培養したら、前回と同様に自然光と紫外線を当てコロニーの状態を観察します。 特に、LB/amp/+プレートにアラビノースを滴下した部分の状態に注目します。
観察が終了したら、すべてのプレートをオートクレーブバックに廃棄します。そのほか菌が付着したものはすべてオートクレーブバックに入れます。
廃棄できないものはエタノールで消毒します。 最後に実験台をエタノールで消毒し、手も消毒します。最後に手をよく洗って終了です。
自然光でのコロニーの色・紫外線照射時のコロニーの色・コロニーの数を記録します。図にはプレートの様子を簡単にスケッチします。またそうなった理由も記入します。
| プレート | 図 | 説明・コロニーの数・考えられる理由 |
LB/− |
 |
|
LB/amp/− |
|
|
| LB/amp/+ | |
|
| LB/amp/+ にアラビノースを滴下した後 |
|
|
| LB/amp/ara/+ | |
| LB/−コントロールプレートとLB/amp/−の結果を確認します。比較することでどんなことがわかりますか。 |
|
| LB/amp/−とLB/amp/+ の結果を比較することでどんなことがわかりますか。 |
|
| LB/amp/+とLB/amp/ara/+ の結果を比較することでどんなことがわかりますか。 |
|
| LB/amp/+にアラビノースを滴下した実験の結果からどんなことがわかりますか。 |
|
| 形質転換が起こる効率を計算したい(形質転換が起こる確率)。今回の実験に加えてどのような実験を行えばよいですか。 |
|